Traditionally, microbial communities that inhabit the human body and surrounding environments have been characterized through culturing on selective plates. However, this approach shows a low sensitivity and does not allow for exploring the unculturable fraction of the microbiome that may account for between 60% and 80% of the observable bacteria.
Although culture-independent methods based on characterizing the 16S ribosomal ribonucleic acid (rRNA) genes and shotgun sequencing of the whole genome have been developed, understanding the complexity of the microbiome in human and non-human environments remains a distant target.
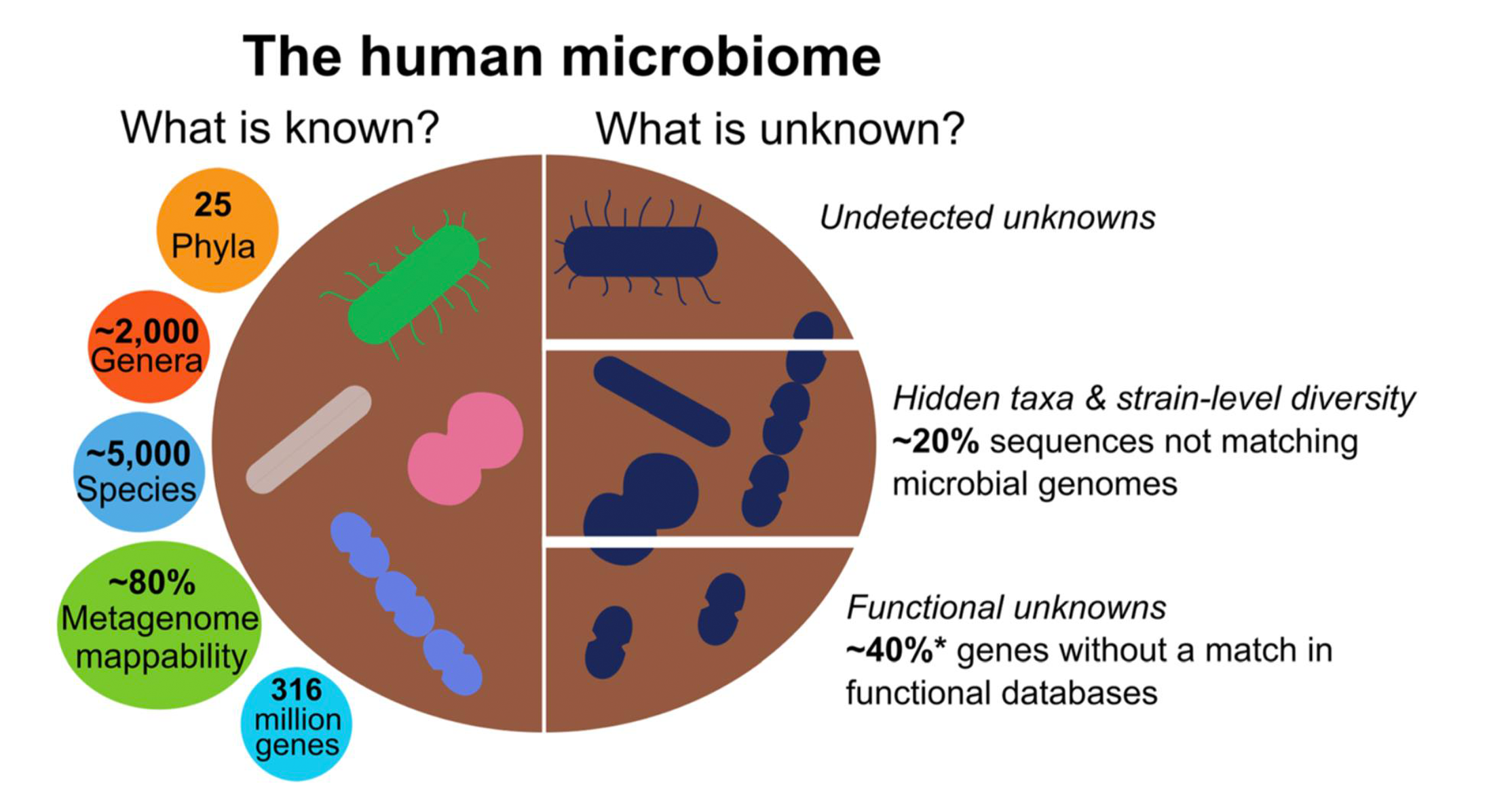
In an attempt to further our knowledge, Andrew Maltez Thomas and Nicola Segata from the University of Trento (Italy) scope out the “unknowns” in contemporary human microbiome research.
Metagenomics has increased the number of microorganism genomes that are mappable up to 85%, which means they can be prioritized for analysis. Based on a recent large-scale metagenomic study of the microbiome across different body sites in individuals with both Westernized and non-Westernized lifestyles, the diversity of the human microbiome has been estimated at 25 phyla, an average 2,000 genera and 5,000 species, and 316 million genes.
However, the diversity of a large fraction of the human microbiome remains unexplored.
There are microbial community members that may be potentially crucial taxa, but which are not easily detectable with cutting-edge metagenomic approaches. For instance, host cells and their DNA in the samples analyzed can hinder the discovery of relevant microbial taxa. Often, eukaryotic microbes and viruses are undersampled by metagenomic protocols. This is the case, for example, with bacteriophages, as reproducible protocols for analyzing them using metagenomics have only recently emerged and are considered relevant colonizers of the human gastrointestinal tract.
The fact that a fraction of genetic material in microbiome species is not yet mappable make it difficult for scientists to characterize hidden taxa and microbiome diversity at strain level. Genes that are only present in a small number of strains or even genes unique to a single strain—also called the variable genome, which is acquired by lateral gene transfer mechanisms—outnumber the genes present in all strains of a species by a ratio of 10 to 1.
Last, but not least, it is difficult to have a complete picture of the functional diversity potential of the human microbiome, mainly due to the inherent nature of high-throughput methodologies that show an imbalanced cost-effective relationship. According to the authors, this is probably the most important caveat in the field.
Thomas and Segata finally cover how emerging technologies will help with dealing with current human microbiome unknowns. Paradigm shifts in human microbiome study may arise from the development and improvement of technologies that include microbial culturomics, single-cell genetic analysis of rare and uncultivated microbes, and in-depth characterization of metagenomic datasets in large-scale populations.
By and large, understanding the functional potential of the microbiome, including the microbial transcriptome, metabolome and proteome, remains the biggest challenge faced by scientists in the field.
Applications of such advancements include a better selection of donor samples for fecal microbiome transplantation, expanding disease-predictive microbiome signatures, and better characterizing of populations and non-human environments, which are not currently subject to extensive study.
Reference:
Thomas AM, Segata N. Multiple levels of the unknown in microbiome research. BMC Biol. 2019; 17(1):48. doi: 10.1186/s12915-019-0667-z.